The energy of the ground state and the excited state (via ΔSCF)
- Hartree-Fock, Kohn-Sham DFT, including LDA,GGA, meta-GGA, range-separated functionals, hybrid functionals, double-hybrid functionals, etc., support dispersion correction

BDF (Beijing Density Functional) is an independent, complete and distinctive quantum chemistry computing software package, especially good at the calculation of the electronic structure of heavy element systems. It is also the world's first fully relativistic density functional program based on modern density functional theory that can accurately calculate the total energy of the ground state of molecular systems (early similar programs could not accurately calculate the total energy due to poor numerical integration accuracy).
The function of the gray background part will be launched in the subsequent version. Stay tuned.
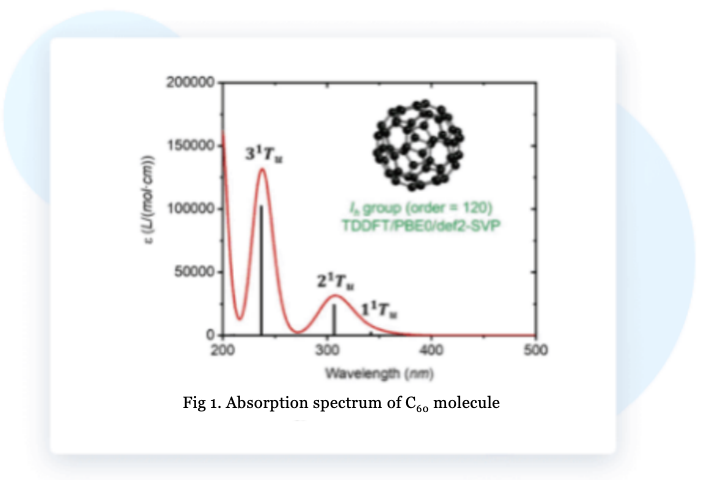
BDF calculates the excitation energy and oscillator strength of the excited state, and then uses the Python script of the BDF program to perform Gaussian broadening of the absorption of the excited state at a certain half-peak width, and finally plots the absorption spectrum of the molecule C₆₀.
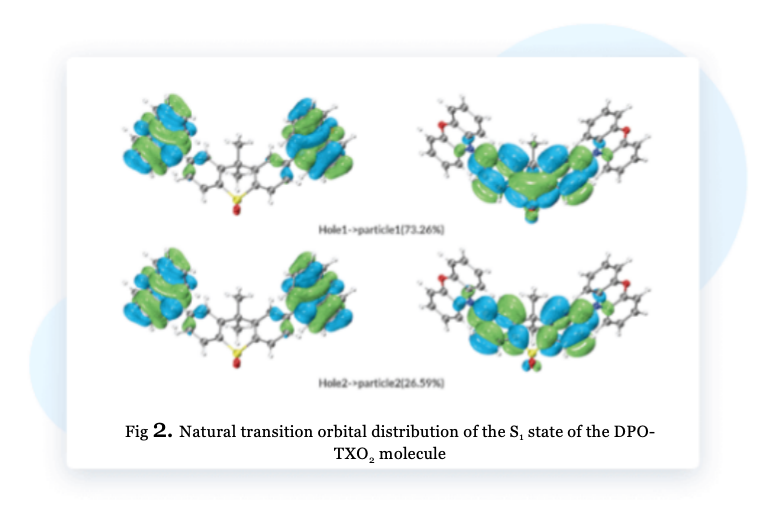
NTO analysis can provide a clearer understanding of the results of excited state transitions. As shown in Figure 2, after NTO analysis of the S₁ state of the DPO-TXO2 molecule, it can be observed that the transition from the occupied orbital Hole1 to the non-occupied orbital particle1 is dominant, contributing 73.26%. The contribution of occupied orbit Hole2 to non-occupied orbit particle2 is 26.59%. The electrons in the excited state of S₁ transition from the electron-donating groups of phenoxazine on both sides to the electron-withdrawing groups at the center.
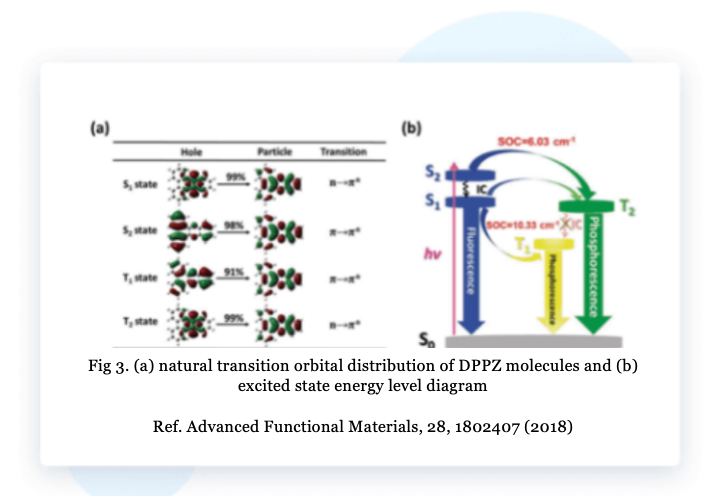
The BDF program calculated the natural transition orbitals (NTO) of DPPZ molecules and the spin-orbit coupling matrix elements between singlet and triplet excited states to analyze the intersystem crossing process. As shown in Figure 3a, the hole distribution of S₁ and T₂ has distinct n orbital characteristics, and the electron distribution has distinct π* orbital characteristics, which are n-π* transitions, while S₂ and T₁ correspond to π-π* transitions. As shown in Figure 3b, intersystem crossing can occur between S₂-T₂ and S₁-T₁.
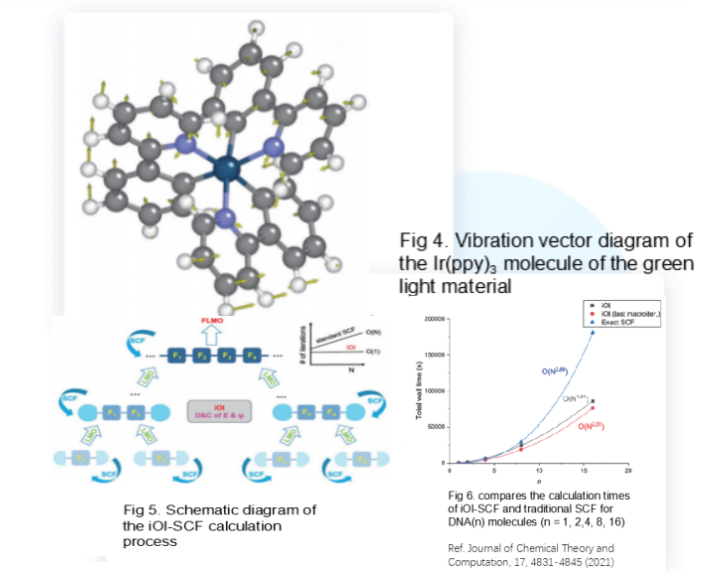
Fig 4. Vibration vector diagram of the Ir(ppy)3 molecule of the green light material
Fig 5. Schematic diagram of the iOI-SCF calculation process
Fig 6. compares the calculation times of iOI-SCF and traditional SCF for DNA(n) molecules (n = 1, 2,4, 8, 16)
Ref. Journal of Chemical Theory and Computation, 17, 4831-4845 (2021)
Fig 7. Schematic diagram of the non-adiabatic coupling calculation process
Ref. Chemical Reviews,121(16),9873-9926(2021)
Fig 8. Schematic diagram of the conical cross calculation process
Ref. M. Boggio-Pasqua, Computational mechanistic photochemistry: the central role of conical intersections. Diss. Universite Toulouse III, 2015
The vibration frequency calculated by the BDF program can be used to determine whether the structure is the minimum point of the potential energy surface (imaginary frequency) and to calculate thermodynamic data.
For large systems (such as those with more than 300 atoms), traditional SCF calculation methods are often no longer applicable. In contrast, the iOI method starts from relatively small molecular sheets, continuously increasing and fusing the molecular sheets until they just reach the required size, and then performs global calculations. iOI-SCF calculation can save a lot of computing time and also avoid the problems of numerous local iterations and easy non-convergence in large systems.
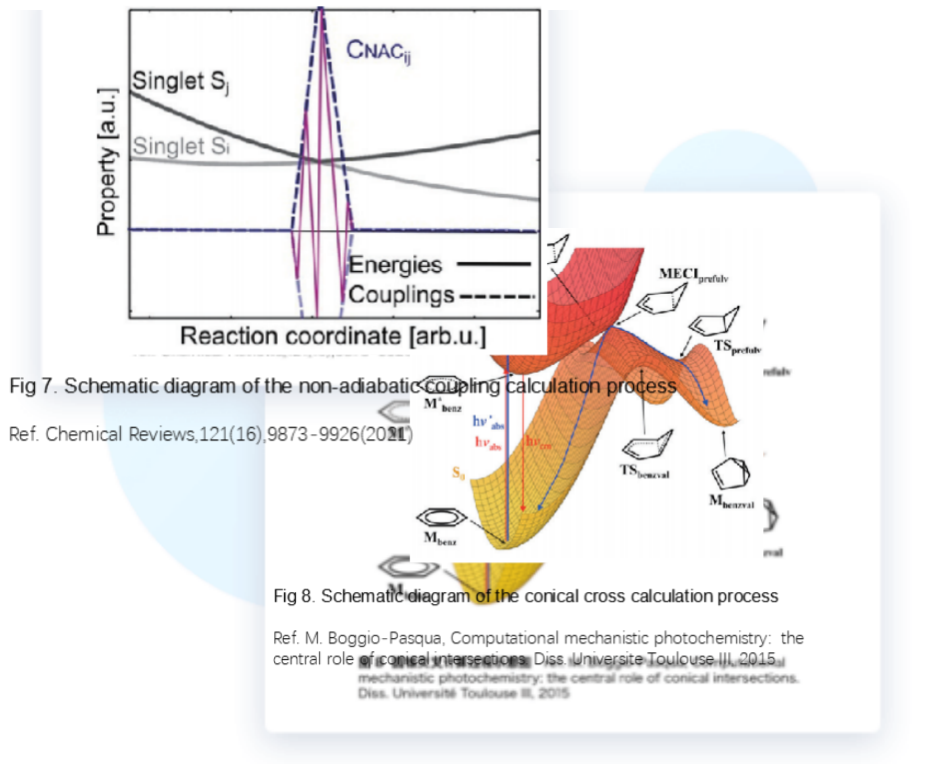
BDF can achieve the calculation of non-adiabatic coupling matrix elements from ground state to excited state and from excited state to excited state. During the non-radiative transition process, the energy of the electronic state and other electronic states may not be effectively separated, and there will be more or less electronic state crossover, leading to the failure of the Born-Oppenheimer approximation. Therefore, the non-adiabatic coupling matrix element is of great significance in the non-radiative transition process.
The BDF program can optimize CI and MECP, that is, the intersection point of the potential energy surfaces of the two states is the conical intersection point, and the point with the lowest energy on this curve of the potential energy surface intersection point is the lowest energy intersection point.
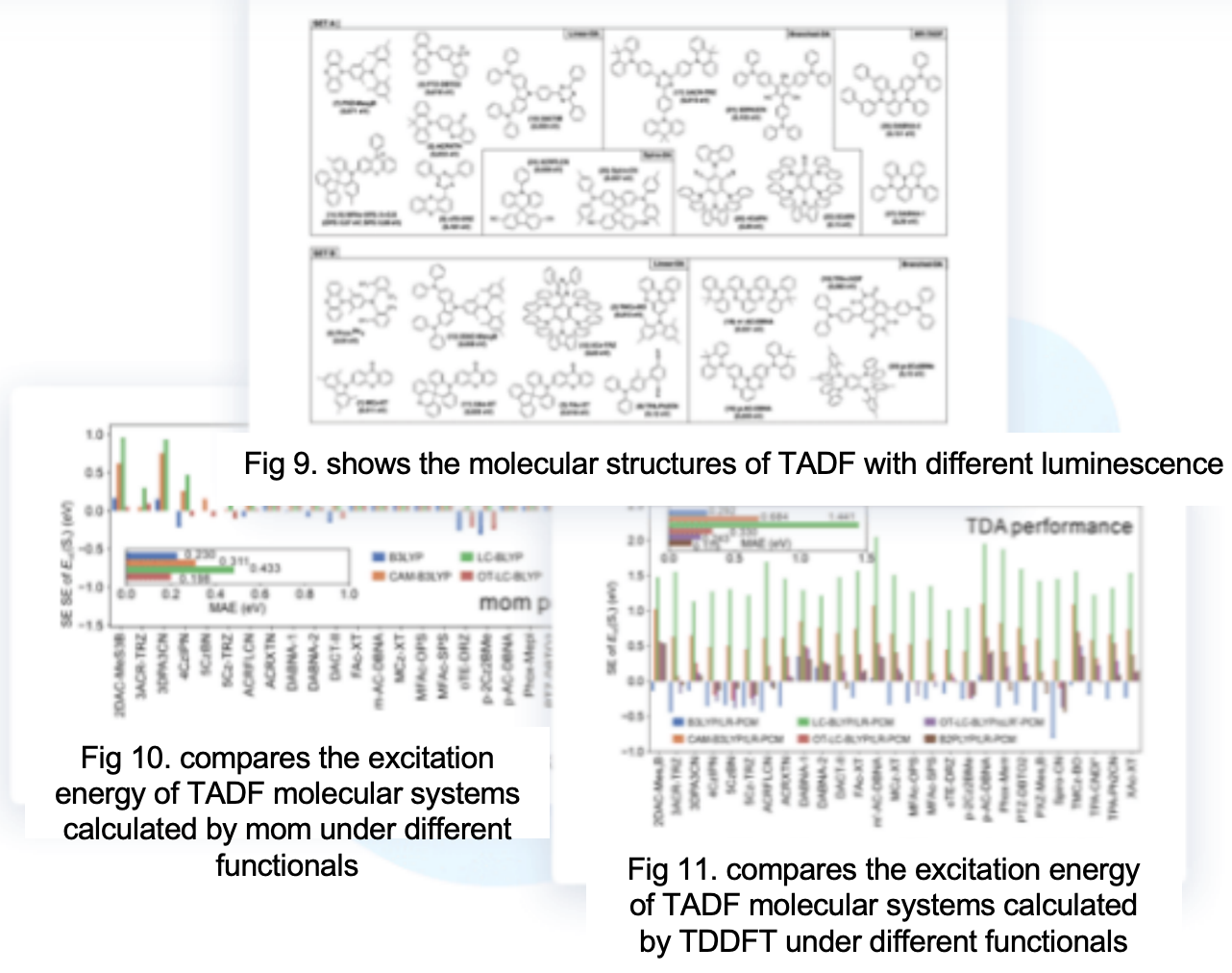
For long-range charge transfer systems, such as typical thermally activated delayed fluorescence (TADF) materials, the TDDFT calculations of pure functionals and global hybrid functionals often tend to underestimate the excitation energy. In contrast, the ASCF method, namely mom, has a smaller calculation error. Combined with functional tuning (optimal-tuning,OT) to reduce the self-interaction error of the functional, the mom method can accurately calculate the S₁ emission energy of TADF molecules.
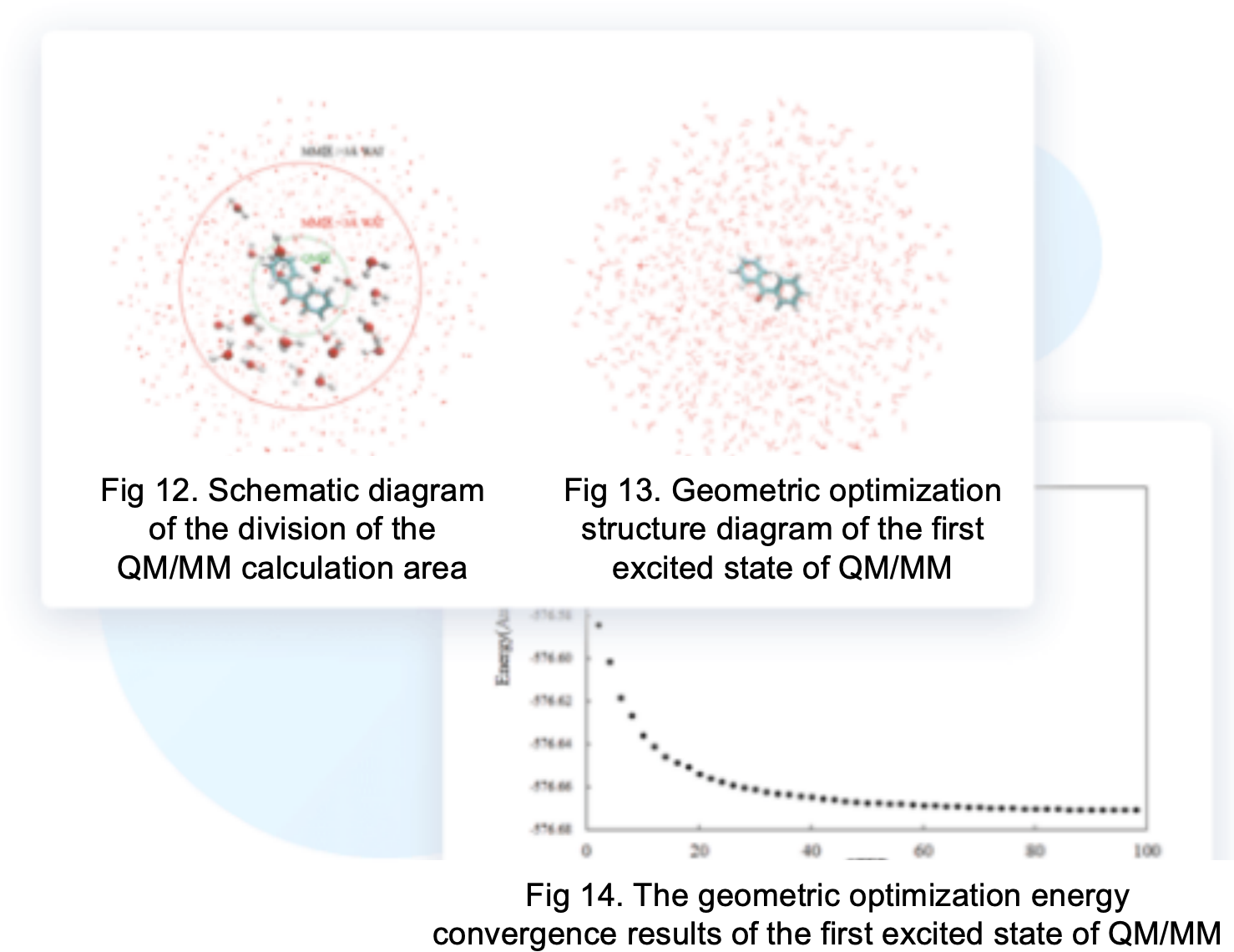
BDF can achieve QM/MM calculations, which not only incorporate the precision of quantum chemistry but also utilize the efficiency of molecular mechanics. Its fundamental idea is to handle the center of interest with quantum mechanics and the rest with classical molecular mechanics.