
DS-PAW is a first-principles calculation software based on plane waves, using pseudopotentials constructed by the Projector Augmented Wave (PAW) method. It is powerful and can perform electronic structure calculations and molecular dynamics simulations, and is widely used in metals, semiconductors, insulators, lithium batteries, biomedicine and other fields.
Main Functions

Exchange-correlation functionals & corrections
- LDAs, GGAs, hybrid functionals, DFT+U correction, vdw-DF functional/DFT-DF correction

Electronic structure
- Density of states, electron localization density, partial charge (pcharge), Bader charge analysis, band structure, band unfolding, Wannier basis

Structural evolution
- Conjugate gradient method, quasi-Newton method, NEB transition state search, BO first-principles molecular dynamics simulation

Magnetic properties
- Linear spin, nonlinear spin, spin-orbit coupling

Linear response theory
- Phonon properties, elastic constants, dielectric properties, Born effective charge, piezoelectric tensor

Optical properties
- Response of dielectric function to frequency

More functions
- External electric field, implicit solvation model, constant potential bias
Software Highlights
Comprehensive Functionalities
- •DS-PAW constructs pseudopotentials using the PAW method. On one hand, by reconstructing the all-electron wave function, the PAW method attains high computational accuracy. On the other hand, being similar in form to the USPP pseudopotential method, the PAW method is straightforward to implement with a moderate consumption of computational resources. Consequently, it achieves an excellent balance between computational accuracy and computational efficiency.
- •DS-PAW determines the ground state of the system by self-consistently solving the Kohn-Sham equations. It supports a variety of exchange-correlation density functionals, including LDAs, GGAs, and hybrid functionals. Moreover, it also supports corrections such as DFT+U, vdw functionals, and DFT-D van der Waals corrections. Based on the self-consistent ground-state solution, various electronic structure properties, optical properties, and potential function distributions of the system can be obtained. By incorporating electron spin, magnetic properties can be computed. In combination with structure evolution algorithms, structure optimization and BO first-principles molecular dynamics simulations can be carried out. Integrating linear response theory allows for the acquisition of phonon properties, elastic constants, dielectric tensors, Born effective charges, and piezoelectric tensors. In future versions, new features will be continuously introduced and optimized in light of user feedback.
User-friendly Interface
- •Leveraging the Hongzhiwei Cloud and Device Studio platform, the onerous process of preparing input files and data processing is obviated. Users can effortlessly complete complex calculations and post - processing tasks with just a few mouse clicks in Device Studio.
- •For users accustomed to the Linux operating system, the current version comes with dspawpy as an auxiliary tool. This tool streamlines the process of preparing input files and enables intricate post - processing and graphical representation.
- •Commonly used computational routines (such as phonon spectrum calculations and Bader charge analyses) are integrated within the software, thereby simplifying user operations.
- • The software parameters are modularly organized, facilitating a direct understanding of the parameter meanings and their interrelationships from the parameter names. The default parameter settings are well - calibrated, enabling novice users to achieve desired functions with minimal parameter adjustments. Additionally, a comprehensive set of adjustable parameters is available to support in - depth calculations for advanced users.
Multi-scenario Applications
- •DS-PAW finds applications in a wide range of scenarios and fields, including lithium - ion battery materials, semiconductor materials, chemical catalysis, alloy design, electrochemical reactions, and the design of emerging materials. The functionality of DS-PAW is designed to align closely with real-world scenarios and fields. In the future, new features tailored to the specific requirements of these scenarios and fields will be continuously incorporated.
Application Case
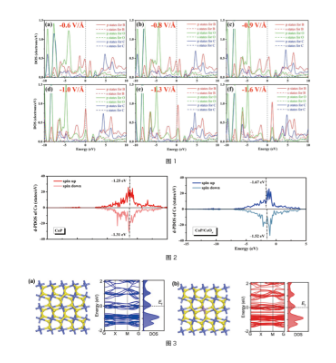
Electronic Structure Analysis
- •
Figure (1): DOS plots of CO₂ adsorbed on the surface of the catalyst B@2DNSe under different electric fields. ref. Physical Chemistry Chemical Physics, 23, 23219 (2021)
- •
Figure (2): PDOS and D-band center plots of CoP and CoP/CeO. ref. Inorganic Chemistry Frontiers, 9, 3047-3058 (2022)
- •
Figure (3): Structure and band plots of CoS₂ and Mn-CoS₂. ref. ACS Applied Materials & Interfaces, 14, 33151-33160 (2022)
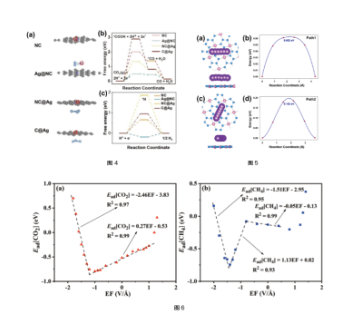
Adsorption Energy, Reaction Energy, Energy Barrier
- •
Figure (4): Structure of different catalysts (a), and CO2RR catalytic reaction free energy (b) and HER free energy (c). ref, ChemElectroChem, 9, e202200987 (2022)
- •
Figure (5): Two diffusion paths (a)(c) and corresponding diffusion energy barrier (b)(d) of K on monolayer BeN. ref. Journal of Alloys and Compounds, 936, 168351 (2023)
- •
Figure (6): Adsorption energy of CO2 (a) and CH at different electric field strengths (EF) (b). ref. Physical Chemistry Chemical Physics, 23, 23219 (2021)
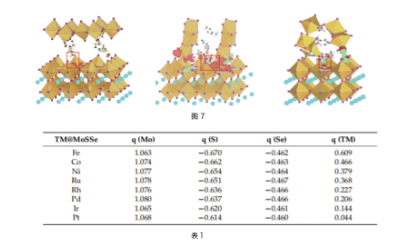
Charge Analysis: Differential Charge Density, Bader Charge Analysis
- •
Figure (7) : Differential charge profiles for different heterojunctions, red represents charge accumulation, cyan represents charge depletion, and red dashed box represents the interaction of organic groups with iodine vacancy defects. ref. Solar RRL, 6, 2200340 (2022)
- •
Table (1) : bader charge analysis table of TM@MoSSe. ref. Molecules, 27, 6038 (2022)
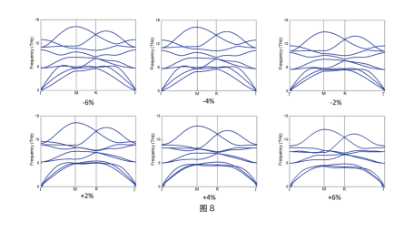
Phonon Spectrum
- •
Figure (8): phonon spectrum of Ca2N at different strains (-6%~6%). ref. Journal of Materials Chemistry A, 10, 12510-12517 (2022)
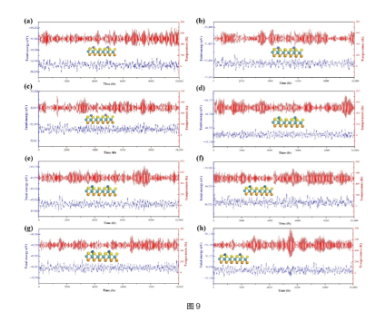
Molecular Dynamics Simulation
- •
Figure (9): Temperature and energy changes over time for AlMD simulations of (a) Fe,(b) Co,(c) Ni,(d) Ru, (e) Rh, (f) Pd,(g) Ir and (h) Pt@MoSSe. Simulations were run at 500 K for 10 ps with a time step of 1 fs. ref. Molecules, 27, 6038 (2022)