Boundary condition
- Periodicity, Dirichlet, and von Neumann open boundaries

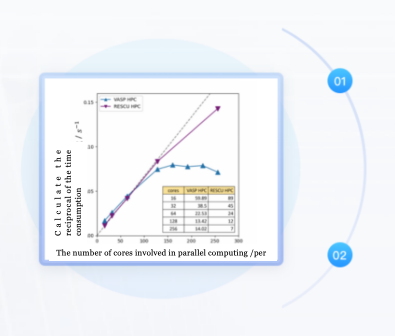
RESCU (Real Space Electronic Structure CalcUlator) is a KS-DFT calculation software that can study very large systems with just a small computer. It combines the respective advantages of plane waves, linear combinations of atomic orbitals, variable-precision meshes, and Chebyshev filtering algorithms. Through optimizations in multiple numerical algorithms, it significantly reduces the burden of traditional DFT calculations while ensuring the accuracy of DFT calculations, making DFT calculations for systems containing tens of thousands of atoms possible. It is a landmark new approach to solving the KS-DFT problem of very large systems. RESCU is currently widely applied in various fields such as metals, semiconductors, insulators, liquids, DNA, 1D, 2D, 3D, surfaces, molecules, magnetic, non-magnetic, impurities, solids, etc.
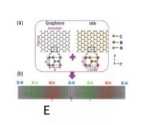
1D Moire pattern simulation
ref. Physical Review Letters, 121(18): 186403(2018)
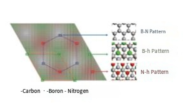
2D Moire pattern simulation
ref. Physical Review Materials, 1(6): 061003(2017)
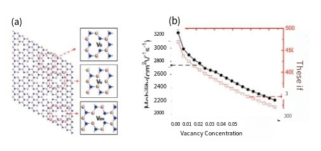
Simulation of carrier mobility at different vacancy concentrations
ref. 2D Materials.4(4 045014(2017)

Simulation of carrier migration in twisted graphene
ret. Physical Review B. 96(19): 195406(2017)
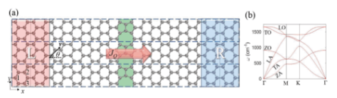
Phonon spectrum calculation
ref. Physical Review B,99(6): 064302 (2019)
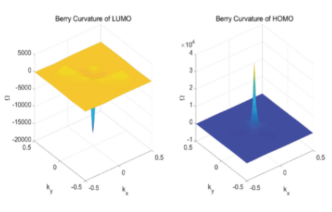
Berry rate calculation
ref. Physica E: Low-dimensional Systems and Nanostructures, 126: 114390(2021)
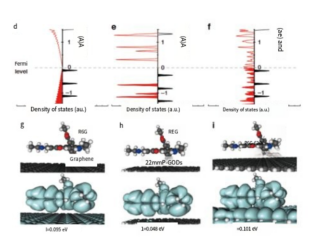
Simulation of density of states and charge transfer
ref. Nature Communications, 9(1): 193(2018)
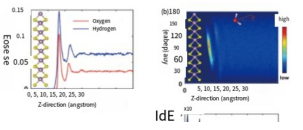
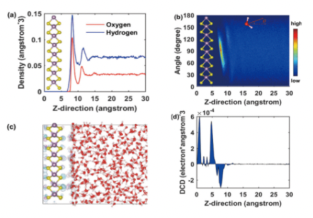
H2O Molecular dipole analysis and differential charge analysis
ref. npj Computational Materials,7(1): 1-9(2021)
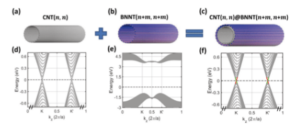
Energy band simulation of carbon nanotubes
ref. Nano Letters, 19(6): 4146-4150(2019)
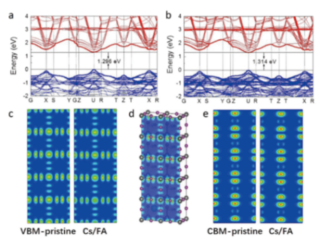
Removal band calculation of low-concentration doped dense Brillouin region
ref. ACS Applied Materials & Interfaces, 11(14): 13812-13821(2019)